
Calliope Dendrou
Dr, Sir Henry Dale Fellow
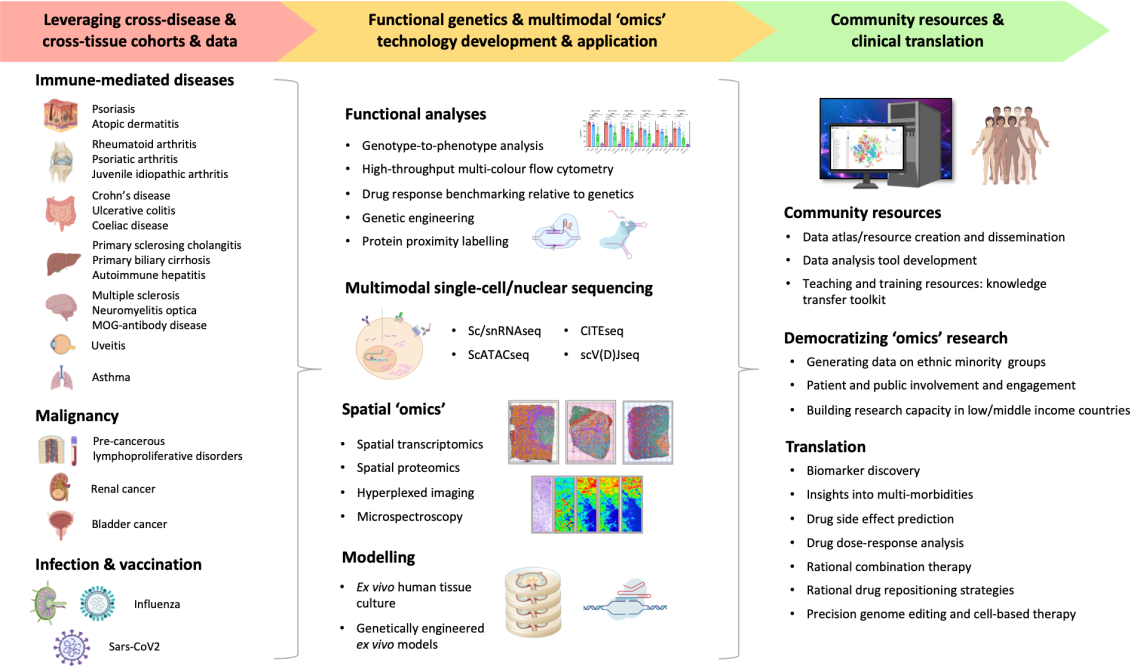
Our research aims to refine and to develop diagnostic, prognostic and therapeutic strategies for improved healthcare provision to patients with immune-mediated and infectious diseases.
We investigate molecular circuits and cellular mechanisms across tissues and across diseases for the purpose of identifying ‘hubs’ that may be targeted therapeutically via drug repositioning approaches. We are also interrogating the relationship between variation in cell responsiveness and transcriptional kinetics and the spectrum of immune-related diseases spanning malignancies, autoimmunity and infections to help inform risk-benefit analyses in the context of therapeutic targeting.
The Dendrou group is supported by the Wellcome Trust, Royal Society, UKRI Medical Research Council, Chan Zuckerberg Initiative, BBSRC, Rosetrees Trust, Diamond Light Source, John Fell Fund, Janssen, Celsius Therapeutics, and Nimbus Therapeutics.
We are highly collaborative – working locally and internationally with experimental and computational scientists and clinicians. Some of our ongoing research programmes include:
(1) Investigating the architecture of genetic predisposition across immune-mediated diseases to explore the functional relevance and potential clinical utility of such cross-comparisons; https://www.nature.com/articles/d41586-021-01839-6
(2) Precision genome editing with tandem autologous transplantation as a therapy for multiple severe immune-mediated diseases; https://gtr.ukri.org/projects?ref=MR%2FT030410%2F1
(3) Creating cellular atlases across multiple immune-mediated and infectious disorders and malignancies through e.g. the Human Cell Atlas, MAP-JAG, TAURUS, and the Oxford-Janssen Cartography Collaboration
(4) Mapping human inflammatory responses in ethnically diverse groups using a novel ex vivo lymph node model; https://chanzuckerberg.com/science/programs-resources/single-cell-biology/inflammation-projects/mapping-human-inflammatory-responses-in-ethnically-diverse-groups/
(5) The Lymph nodE single-cell Genomics AnCestrY (LEGACY) Network: contextualizing the vaccine response of lymph nodes through the creation of an ancestrally diverse single-cell atlas; https://www.well.ox.ac.uk/news/legacy-network-receives-2million-in-funding-from-the-chan-zuckerberg-initiative-for-single-cell-ancestry-vaccine-research
Recent publications
Identification of genetic risk loci associated with aquaporin 4-positive neuromyelitis optica spectrum disorder: a genome-wide association study
Journal article
Attfield KE. et al, (2026), The Lancet Neurology, 25, 482 - 491
Protocol for precision cutting and short-term culture of lymph node tissue slices for modeling of innate immune responses ex vivo.
Journal article
Fergusson JR. et al, (2026), STAR protocols, 7
Early lymph node T follicular helper cell signalling hub drives influenza vaccine response in an ancestrally diverse cohort
Journal article
Siu JHY. et al, (2025), eBioMedicine, 122, 106036 - 106036
Navigating the Hepatic Immune Landscape With Fine Needle Aspiration of the Liver-An Emerging Technique.
Journal article
Lynch KD. et al, (2025), Liver international : official journal of the International Association for the Study of the Liver, 45
Synovial matrix turnover controls immune cell spatial patterning in inflammation resolution.
Journal article
Richard J-B. et al, (2025), Molecular systems biology, 21, 1638 - 1665