
Contact information
geraldine.gillespie@ndm.ox.ac.uk
 https://orcid.org/0000-0002-1075-870X
https://orcid.org/0000-0002-1075-870X
Old Road Campus Research Building
Geraldine Gillespie
Professor of Molecular Immunology
- Principal Investigator
- Group Head / PI
- Member of Congregation and Supervisor
- NDM EDI Committee Chair
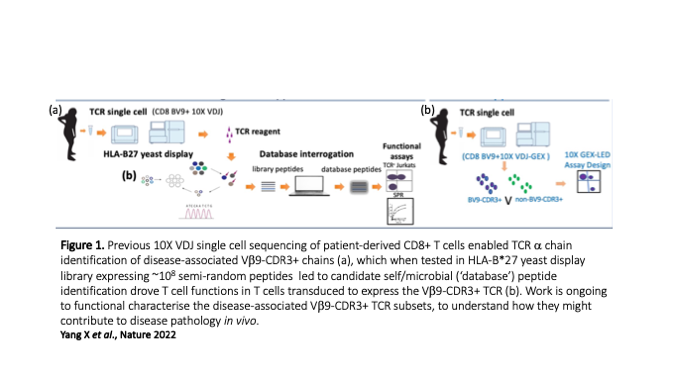
One theme in our group is to understand how specific HLA-B*27 subtypes pre-dispose to inflammatory autoimmune condition, most notably Ankylosing Spondylitis (AS), a condition that affects the spine and specific joints. This association has been recognised for over 5 decades, but the biological basis of disease initiation is not well understood. Through international collaboration, we recently re-explored an original disease model suggesting that a case of mistaken identity (autoimmunity) contributes to disease pathology where CD8+ T cells initially triggered by microbial epitopes bound to HLA-B*27 subsequently attack HLA-B*27-bound to self-peptides. We addressed this by testing a HLA-B*27 yeast display platform expressing large numbers (~108) of random peptides which we then tested for binding to AS-enriched T cell receptors (TCRs). This strategy led to the identification of peptides with sequences that are shared between microbial and self-proteins, adding strong support to the autoimmune disease hypothesis. These T cells have distinct transcriptional profiles that related to inflammatory CD8+ subsets described in other autoimmune conditions, and we next aim to interrogate these cellular phenotypes further to understand how the inflammatory environment modulates the functions, both spatially and temporally. We also plan to interrogate the gut-joint axis to understand the phenotypes and trajectories of T cells at these different sites. In addition to deciphering the complex biology of AS, a goal of this work is to elucidate if these finding could underpin the development of targeted therapies for AS.
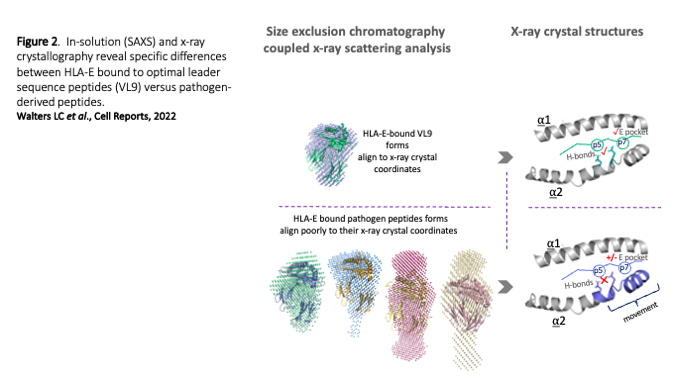
Our second theme follows the unprecedented success of a CMV-SIV vaccine in rhesus macaques that elicited protective, SIV-specific CD8+ T cells targeting multiple peptides restricted by the MHC-E, has stemmed considerable interest in HLA-E (human MHC-E), a non-classical MHC protein originally described in the context of NK cell regulation and function. Unlike classical MHC class I, HLA-E proteins are near monomorphic and therefore offer a ‘universally’-conserved T cell restriction element. Our recent HLA-E structural studies have revealed the molecular basis underlying HLA-E-peptide binding and our biochemical studies have revealed unusual HLA-E properties that might explain how HLA-E picks up peptide epitopes in vivo. This work has also enabled us to improve HLA-E ELISA-based screening methods to quantitate peptide binding and to generated improved HLA-E tetramer tools to study HLA-E restricted T cells. With collaborators we aim to develop therapeutic reagents (primarily antibodies that recognise HLA-E-peptide antigen) targeting both viruses such as HIV in addition to tumour specific/associated antigens (e.g. from AML or HPV+ tumours). Our goals are to ultimately develop such reagents as CAR T cell-based therapeutics, alongside T cell receptor-based therapies, in addition to devising methods to create novel HLA-E targeting vaccine platforms.
Recent publications
Generation of T cells with reduced off-target cross-reactivities by engineering co-signalling receptors
Journal article
Cabezas-Caballero J. et al, (2026), Nature Biomedical Engineering, 10, 753 - 764
Targeting MHC-E as a new strategy for vaccines and immunotherapeutics
Journal article
Früh K. et al, (2026), Nature Reviews Immunology, 26, 52 - 66
Shared HLA-E and Mamu-E Peptide Repertoires With Subtle Peptide Binding Differences Revealed by Combined nDSF- and Fluorescence Polarisation-Based Methods.
Journal article
Quastel MN. et al, (2026), European journal of immunology, 56
A cytoplasmic motif in HLA-E that drives clathrin-mediated endocytosis and VCP-associated postendocytic trafficking
Journal article
He W. et al, (2025), Proceedings of the National Academy of Sciences, 122
HLA-E: Immune Receptor Functional Mechanisms Revealed by Structural Studies.
Journal article
Gillespie GM. et al, (2025), Immunological reviews, 329
Identification and structural characterization of a mutant KRAS‐G12V specific TCR restricted by HLA‐A3
Journal article
Sim MJW. et al, (2024), European Journal of Immunology, 54
HLA-E-VL9 antibodies enhance NK cell and CD8 + T cell cytotoxicity against HIV-infected CD4 + T cells
Preprint
Hwang JK. et al, (2024)
Mucosal signatures of pathogenic T cells in HLA-B*27+ anterior uveitis and axial spondyloarthritis.
Journal article
Paley MA. et al, (2024), JCI insight, 9